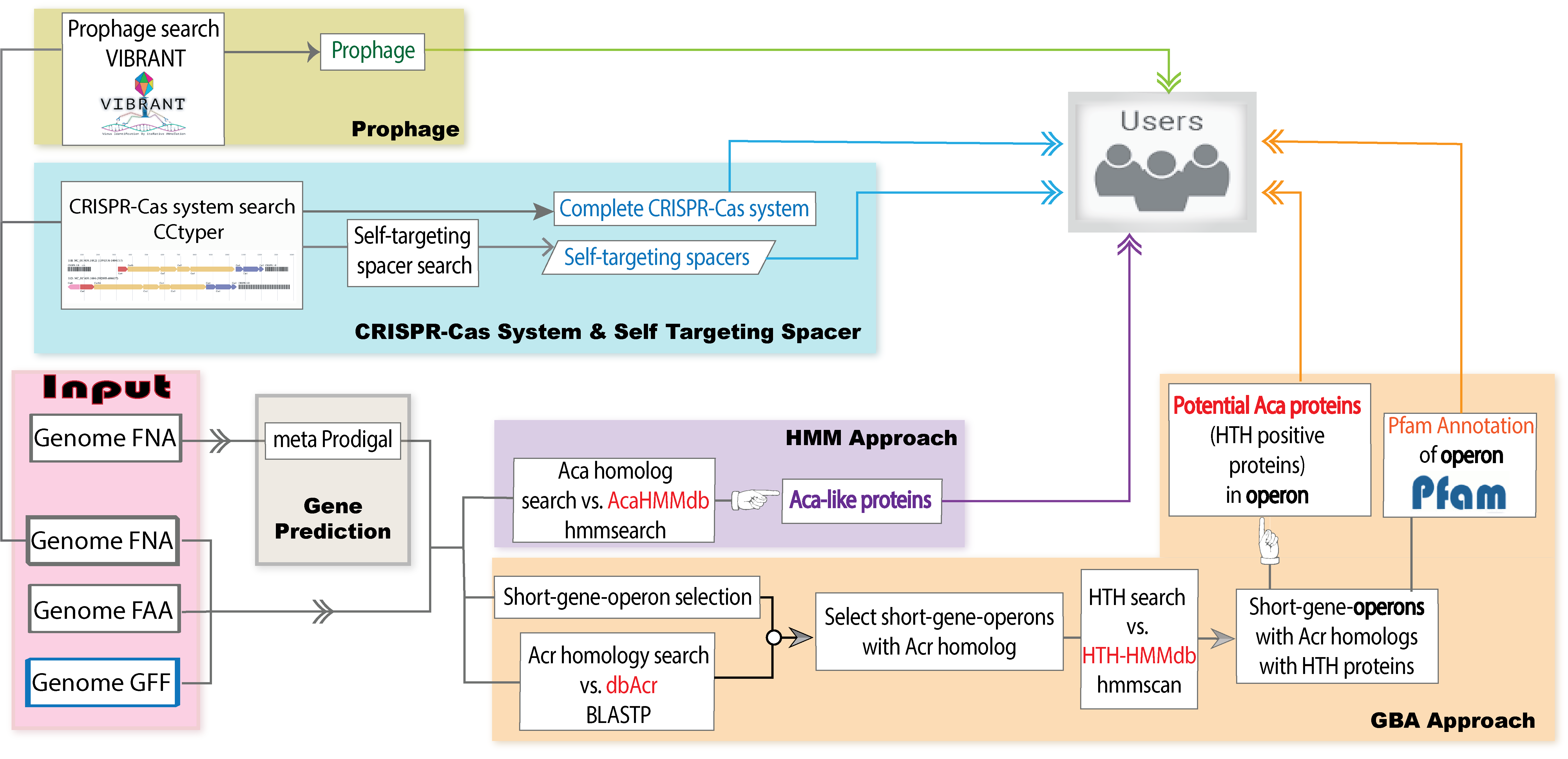
AcaFinder is the first ever automated genome mining tool for reliable
Acas screening. To more confidently identify Acas given a genome or metagenome assembled genome, AcaFinder implementes two approaches. The first approach is based on guilt-by-association (GBA), meaning that we identify homologs of Acrs first and then search for
HTH-containing proteins in the acr gene neighborhood. The second approach is to build an HMM (hidden markov model) database using training data of the
12 known Aca families, and then search for Aca homologs with this Aca-HMMdb instead of Pfam HTH HMMs. In addition to the two implemented approaches, AcaFinder also integrates a CRISPR-Cas search tool (
CRISPRCasTyper), a prophage search tool (
VIBRANT), and in-house a Self-targeting spacer (STSS) searching tool, providing users with detailed information vital to the assessment of Aca predictions.